Décrypthon : de la bio-informatique appliquée
Le problème scientifique
L’idée du calcul est basée sur le raisonnement suivant : si le « codage » de deux protéines est analogue, alors formuler l’hypothèse que leurs fonctions présentent des analogies est une hypothèse fréquemment plausible.
Le cahier des charges informatique : la théorie
Il s’agit de comparer deux à deux les enregistrements d’une base de données :
-
la base de données
Il est admis qu’à chaque protéine connue est associée une séquence peptidique définissant sa structure chimique. La base de données est donc constituée de toutes les séquences des protéines connues. -
le critère mathématique
Un critère de similarité entre deux séquences est défini par la mesure du plus petit nombre d’opérations qui, appliquées à une séquence, permettent d’obtenir la seconde ; intuitivement on peut interpréter cette distance d’édition comme une mesure du nombre d’événements survenus au cours de l’évolution entre deux séquences et leur plus récent ancêtre commun.
Le cahier des charges informatique : la pratique
En fait la base de données des séquences connues n’existe pas en pratique.
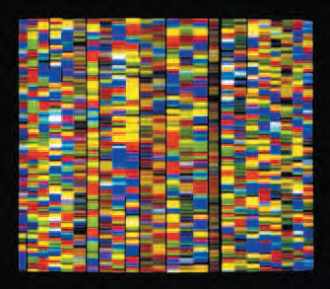
Visualisation des séquences d’ADN dans le but de décrypter le génome humain.
© INSERM, PHOTO JORDAN B. DR/HUNKAPILLER M.
Il existe plusieurs « grandes » sources de référence, d’accès public ou privé, mais, et c’est l’une des difficultés de base de la bio-informatique, aucune n’est à aucun moment exhaustive ou de format standardisé. Elles ont des intersections souvent non vides et parfois denses, et ce sont des sources de formats hétérogènes. Aussi, dans le contexte de la « bio-informatique », la première étape du cahier des charges consiste à fabriquer une base de données homogène et non redondante à partir de plusieurs sources hétérogènes et redondantes. Dans le cas du Décrypthon1, la compilation initiale de quatre grandes bases de données et de 80 protéomes entièrement séquencés générait 1 200 000 séquences : par élimination des redondances exactes (séquences parfaitement identiques), cet ensemble a été réduit à 560 000 séquences/
Pour ce volume de comparaison, l’évaluation du calcul représente 15 millions d’heures : soit plus de mille cinq cents ans sur un seul PC, ou un an de calcul à plein temps sur 1 500 PC en cluster ! Compte tenu de la dimension des calculs envisagés, la réalisation du calcul proprement dit a constitué une première en France.
Lors du « Téléthon » de décembre 2001 un appel à volontaires a été lancé pour mettre à disposition du temps machine de leur ordinateur personnel. Le calcul envisagé, 150 milliards de comparaisons à réaliser, se prête bien en effet à des découpages en paquets. En février 2002, 75 000 volontaires ont donc pu télécharger par Internet le programme de comparaison.
Le résultat final
L’opération ayant été rendue possible grâce à l’aide des bénévoles, l’ensemble des résultats a été très rapidement mis à la disposition de la communauté scientifique sur un site public d’accès libre :
http://infobiogen.fr/services/decrypthon
Utilisations possibles
Parmi quelques types d’exploitation possibles : les familles de protéines
Genomining est une société de bio-informatique fondée en mai 2001 par William Saurin, normalien, directeur de Recherches au CNRS, et Laurent Voignac (79), ingénieur en chef des Mines.
Genomining a été en 2002 l’opérateur scientifique du Décrypthon mené en partenariat avec l’AFM et IBM.
www.genomining.com
Une question scientifique fondamentale est de pouvoir rassembler les protéines en « familles », dont chaque membre posséderait donc un « point en commun » avec les autres membres. Le problème mathématique de rassembler des objets en familles si on connaît le degré de ressemblance des objets deux à deux n’est pas simple. Un critère de transitivité appliqué trop brutalement peut conduire à rassembler tous les objets dans une seule grande famille, ce qui apporte peu d’information. À l’inverse un critère trop strict conduit à un éparpillement de familles, à l’extrême les familles ne comportent qu’un objet.
Dans le monde de la biologie la perspective de rassembler les protéines en familles non triviales est importante : si l’on peut tenter l’analogie, elle correspondrait à rassembler les éléments chimiques en colonnes du tableau périodique, ce qui permet de prédire des propriétés chimiques communes aux éléments qui partagent la même colonne.
Des travaux théoriques peuvent donc être entrepris pour identifier des familles pertinentes de protéines à partir des informations disponibles sur leur analogie, et de pouvoir en déduire à l’étape ultérieure des propriétés biologiques communes. Ces travaux s’appuyant sur la bio-informatique mêlent des compétences de biologie, de statistique et d’informatique théorique.
Suites
La mise à jour de cette base à partir des protéines nouvellement connues entre janvier 2002 et janvier 2003, soit en un an, devrait nécessiter le même volume de calcul ! La mise à disposition des données des grands projets de séquençage n’est évidemment pas étrangère à cette croissance rapide.
____________________________
1. Les références en volume sont à considérer en janvier 2002, date du début du calcul Décrypthon.
Elles évoluent sensiblement avec le temps : doublement en un an.