Entre physique et biologie déchiffrer le mouvement des protéines

Observer le mouvement des protéines, c’est suivre le détail du fonctionnement de la machinerie cellulaire. Avec la thèse présentée ici, on contribue à la construction d’une méthodologie robuste qui permettra aux chercheurs de tirer les conclusions les plus exactes possibles de leurs observations, aussi bien pour la mise au point de médicaments que pour des expériences de biologie cellulaire.
Selon les problématiques abordées, le biologiste s’intéressera tantôt aux propriétés d’un être tout entier, d’un organe, d’un embryon, d’un amas de cellules, voire encore d’une simple cellule, d’une de ses sous-parties ou même à la chimie des molécules qui constituent la matière biologique. Bien qu’entremêlées les unes aux autres, ces différentes échelles du vivant requièrent chacune l’utilisation de méthodes expérimentales et d’outils d’analyse adaptés.
Au cours de ma thèse, j’ai développé une nouvelle méthode d’analyse du mouvement des protéines, dont l’échelle caractéristique se situe entre celle de la cellule et celle de la molécule : plus « macro » que l’échelle de la chimie, mais suffisamment petite pour que les phénomènes aléatoires dus à l’agitation thermique y soient prépondérants – une échelle pour laquelle la physique et les statistiques offrent de précieuses clés de compréhension. J’ai notamment utilisé cette méthode pour aider Sanofi à valider une nouvelle immunothérapie et pour analyser les expériences d’un chercheur s’intéressant aux origines moléculaires de la maladie de Parkinson.
En illustration : Vue d’artiste de l’intérieur d’une cellule. Les éléments colorés sont des protéines. « Digital Renders by Evan Ingersoll & Gael McGill Paintings by David Goodsell ».
Le mouvement aléatoire des protéines
Pour comprendre l’intérêt et la complexité de l’étude du mouvement des protéines dans les cellules, il est bon d’avoir à l’esprit quelques ordres de grandeur. Tout d’abord, le diamètre typique d’une cellule est de quelques micromètres. Un niveau plus petit, l’ordre de grandeur de la taille des organelles, c’est-à-dire des compartiments ou des unités fonctionnelles d’une cellule, est le micromètre. Pour comprendre le fonctionnement de ces dernières, on peut notamment s’intéresser à leurs principaux composants « actifs » : les protéines.
Ces grosses molécules sont les petites mains de la cellule, ce sont elles qui consomment l’énergie, régulent les flux, transmettent les signaux, etc. Elles mesurent quelques nanomètres de diamètre et sont percutées en permanence par les molécules d’eau qui les entourent, ce qui rend leur mouvement erratique – il est dit « diffusif ». En quelques secondes, au gré de ces fluctuations aléatoires, une protéine peut ainsi traverser la cellule dans laquelle elle se trouve. Cependant, les propriétés dynamiques d’une même protéine peuvent varier considérablement au cours du temps, selon sa géométrie, qui détermine notamment sa propension à se lier avec d’autres, ou selon l’endroit où elle se trouve : enchevêtrée dans une zone peuplée d’obstacles, elle sera certainement moins mobile.
Un désordre bien organisé
Le mouvement d’une protéine semble donc, à première vue, en grande partie gouverné par le hasard. Pourtant, la machinerie cellulaire, pour remplir ses fonctions, ne saurait évidemment pas se contenter d’un désordre « homogène » : chaque protéine doit être acheminée là où elle a son rôle, de manière à ce que des sous-unités fonctionnelles apparaissent au sein de la cellule. Par exemple, les canaux responsables de l’équilibre des ions entre l’intérieur et l’extérieur de la cellule sont composés de plusieurs protéines différentes, qui doivent être localisées au même endroit pour que le canal puisse être assemblé.
De même, les récepteurs chargés de détecter la présence de l’une ou l’autre molécule dans l’environnement de la cellule doivent être conduits à la membrane pour remplir leur fonction. En fait, la dynamique de diffusion est intimement reliée à la fonction des protéines et est donc susceptible de varier grandement d’un type de protéine à l’autre. Ainsi, puisque toutes les protéines n’ont pas les mêmes propriétés de diffusion aux mêmes endroits, certaines prédominent là où d’autres se font rares ; d’autres encore sont confinées à certaines régions.
Certains médicaments affectent le mouvement des protéines
Pendant ma thèse, je me suis attaché à travailler sur des exemples d’applications, pour lesquels il était important de comprendre le mouvement de ces si petits composants de la cellule. J’ai notamment travaillé avec une équipe de recherche de Sanofi chargée d’évaluer les molécules en développement et de sélectionner les meilleurs candidats. Le projet auquel ont contribué mes travaux portait sur la mise au point de nouvelles immunothérapies contre le cancer, c’est-à-dire de traitements visant à stimuler les cellules T (qui sont au cœur de la réponse immunitaire) pour qu’elles « s’activent » et attaquent les cellules cancéreuses.
Plus précisément, je me suis intéressé au mouvement de récepteurs connus pour être capables de déclencher ou d’atténuer l’activation des cellules T. Situées à la membrane, et donc en contact à la fois avec l’intérieur et avec l’extérieur de la cellule, ces protéines enclenchent un mécanisme dans la cellule lorsque se fixent à leur partie externe des anticorps présents dans l’environnement de la cellule. En temps normal, ces anticorps sont produits par les cellules de l’organisme qui dysfonctionnent, pour signaler leur état aux cellules T voisines afin d’être éliminées ; mais certaines cellules cancéreuses bloquent cette signalisation, passent donc inaperçues et prolifèrent. Les immunothérapies visent à contrer cette stratégie en introduisant de façon artificielle des anticorps capables, en quelque sorte, d’appuyer sur l’interrupteur.
Observer l’action des anticorps
Les différents mécanismes d’activation sont encore assez méconnus, et l’on sait notamment peu de chose de leurs toutes premières étapes, qui suivent la détection des anticorps. Quel que soit l’effet de l’anticorps sur le récepteur auquel il se lie, il est probable que le mouvement de ce dernier s’en trouve affecté, de façon plus ou moins marquée : il a été observé dans certains cas que les récepteurs s’immobilisent complètement, mais le changement n’est pas toujours aussi marqué. Au cours d’expériences, nous avons cherché à observer les éventuels changements de dynamique induits par les anticorps candidats, pour caractériser d’éventuelles variations d’intensité, voire de nature, dans les effets produits sur le mouvement du récepteur.
Pour mesurer son mouvement, nous avons marqué le récepteur afin de pouvoir l’observer et avons filmé son déplacement dans des cellules activées avec chacun des différents anticorps. L’objet de ma thèse a été de développer une méthode pour comparer les trajectoires ainsi observées et détecter d’éventuelles différences. Avant d’être ainsi évalués, les anticorps sont bien sûr testés d’autres manières : on sait par exemple qu’ils sont capables, dans des conditions bien précises, de se lier à leur cible. Mais les expériences qui le prouvent sont effectuées dans des milieux très simplifiés, loin de reproduire toute la complexité d’une cellule.
“Les protéines sont percutées par les molécules d’eau, ce qui rend leur mouvement erratique.”
Ainsi, il est possible qu’un anticorps se fixe bien à sa cible dans un tube à essai, mais en soit incapable lorsque ce récepteur est situé à la membrane d’une cellule, parce que des contraintes géométriques l’empêchent de se lier de la même façon ou bien parce qu’il se lie mieux à une autre protéine présente à la surface de la cellule, qui le détournera de sa cible initiale. Il existe d’autres moyens de vérifier que les anticorps ont bien provoqué le stimulus attendu, par exemple en mesurant la concentration de certaines protéines sécrétées par les cellules lorsqu’elles sont activées.
Mais mon travail de thèse permet d’observer une étape intermédiaire, elle donne accès à un nouveau niveau de détail sur le mode d’action des anticorps sur leur cible. Au-delà de la comparaison de molécules candidates, l’analyse du mouvement des protéines est un outil générique qui permet, selon le protocole expérimental choisi, une meilleure compréhension de leur rôle au sein de la cellule et de la manière dont ce mouvement participe aux différentes fonctions dans lesquelles les protéines sont impliquées.
Décrire des trajectoires très diverses
L’exploitation des observations de trajectoires de protéines n’en est pourtant qu’à ses balbutiements : plusieurs difficultés compliquent en effet l’analyse, et je me suis attaché durant cette thèse à construire une méthodologie qui y soit la moins vulnérable possible. La première difficulté est d’identifier un « modèle » générique, dont les trajectoires seraient une réalisation et dont on s’attachera à déterminer les paramètres. Un modèle simple de mouvement aléatoire est celui du mouvement brownien, dans lequel chaque déplacement est indépendant des précédents, suivant une distribution centrée dont la variance constitue le seul paramètre du modèle.
Bien qu’il soit une bonne première approximation, celui-ci est loin de capturer toute la complexité du mouvement des protéines dans les cellules : comme nous l’avons vu plus haut, leur dynamique change en fonction de la région de la cellule dans laquelle elles se trouvent. Heureusement, les physiciens n’avaient pas attendu de pouvoir observer le mouvement de protéines pour s’intéresser aux marches aléatoires : celles-ci ont, entre autres applications, été étudiées dès le siècle dernier pour mieux comprendre le mouvement des électrons dans les métaux, qui détermine leurs propriétés de conduction. Plusieurs modèles de marches aléatoires, prenant chacun en compte un type de « déviation » du modèle central qu’est le mouvement brownien, ont donc été imaginés.
Une méthode qui emprunte à plusieurs modèles physiques
Pour que les propriétés du mouvement soient calculables analytiquement, ces modèles sont restés relativement simples, ou en tout cas trop simples pour qu’on puisse raisonnablement les estimer capables de rendre compte de toute la complexité du mouvement des protéines dans les cellules. Il est plus raisonnable de s’attendre à ce que plusieurs des effets que modélisent les types de marches aléatoires « classiques » agissent simultanément sur la diffusion des protéines dans les cellules. C’est pourquoi la méthode que j’ai développée permet d’interpoler entre plusieurs de ces modèles.
Par ailleurs, comme dans pratiquement tous les cas d’analyse de données, il est nécessaire de définir un niveau de granularité adapté à la quantité et à la qualité des observations dont on dispose. Les conditions expérimentales font que la quantité de trajectoires observées varie de plusieurs ordres de grandeur selon les expériences, le type de protéine observé, etc. Ma méthode associe un modèle à un ensemble de trajectoires : plus l’ensemble est grand, plus le modèle sera déterminé avec précision, mais une centaine de trajectoires donnent souvent une bonne première approximation et la granularité peut donc être ajustée en conséquence : si l’on dispose de milliers de trajectoires par cellule, on pourra comparer entre elles les différentes régions de la cellule. Cependant, si l’on n’en observe qu’une centaine par cellule, on pourra tout de même ne considérer qu’un ensemble par cellule et utiliser ma méthode pour comparer entre elles ces dernières.
Un algorithme entraîné sur des simulations
Au début de ma thèse, j’ai participé à un concours scientifique – une manière de susciter beaucoup d’émulation au sein d’une communauté de chercheurs et de comparer les méthodes proposées selon des métriques communes – dont les résultats ont montré que les réseaux de neurones sont aujourd’hui les meilleurs algorithmes connus pour estimer les paramètres de diffusion. J’ai donc choisi, pour caractériser les trajectoires, d’utiliser un tel réseau, c’est-à-dire une fonction paramétrable dont les coefficients sont ajustés au cours d’une phase d’apprentissage (appelée aussi « entraînement »).
“L’analyse du mouvement des protéines permet une meilleure compréhension de leur rôle au sein de la cellule.”
Ces outils sont largement utilisés pour l’analyse d’images et ce sont eux qui ont tiré le développement de l’intelligence artificielle ces dernières décennies. Cependant, leur entraînement requiert souvent d’importantes quantités de données : par exemple, les algorithmes qui se proposent d’aider les radiologues à identifier les zones les plus intéressantes sur une radiographie nécessitent de disposer de milliers d’exemples préalablement annotés. Ici, on a l’avantage de pouvoir créer à la fois les données et les annotations : puisque la physique des marches aléatoires est assez bien connue, il est possible de simuler des trajectoires dont on connaît les paramètres de diffusion.
Ainsi, en utilisant uniquement des trajectoires simulées, j’ai entraîné un réseau de neurones à inférer les paramètres de diffusion et ai pu l’utiliser pour analyser des trajectoires de protéines observées dans les cellules. La condition pour que cette approche fonctionne est que les trajectoires simulées à l’entraînement « ressemblent » assez aux trajectoires observées. Je me suis donc assuré que les échelles de temps et d’espace correspondent, et j’ai utilisé plusieurs types de marches aléatoires à l’entraînement, de façon à couvrir une grande variété de dynamiques de diffusion.
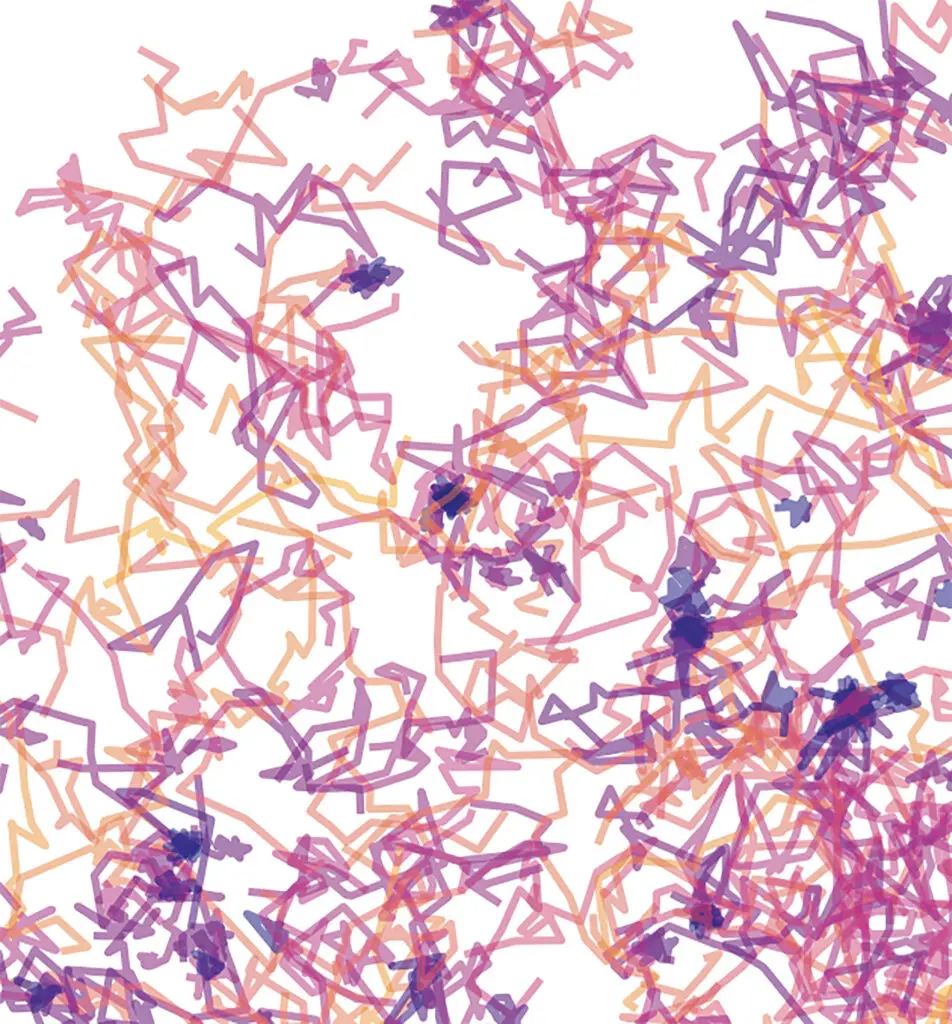
Décrire les trajectoires avec un réseau de neurones
Pour produire l’estimation qu’on l’a entraîné à fournir, un réseau de neurones applique à la trajectoire une série d’opérations. Le plus souvent, seul importe le résultat de la dernière opération, qui constitue la sortie du réseau. Dans notre cas, il s’agit d’estimations de deux paramètres physiques couramment utilisés pour décrire les marches aléatoires. Mais, si l’on regarde quelques étapes en amont, il est possible de « décomposer » l’estimation en plusieurs facteurs, un peu comme plusieurs axes d’une carte sur laquelle le réseau placerait chaque trajectoire.
Pendant son apprentissage, le réseau construit des axes de plus en plus pertinents, et calcule sa prédiction à partir de la position des trajectoires sur cette carte. Appelée « espace latent », cette représentation n’est pas directement interprétable, parce que le réseau n’explique pas à quoi correspond chaque dimension ; néanmoins, elle contient plus d’informations sur la trajectoire que la simple sortie du réseau – elle a été optimisée pour encapsuler au mieux l’information qui lui permet d’estimer ensuite des grandeurs physiques.
J’ai donc choisi d’utiliser l’espace latent pour comparer les trajectoires : ma méthode consiste en fait à comparer les distributions dans l’espace latent issues de deux ensembles de trajectoires, afin d’estimer si elles présentent des différences statistiquement significatives. Cela permet, plutôt que de se concentrer sur un critère préétabli, de comparer les trajectoires selon plusieurs aspects en même temps, ce qui est particulièrement utile lorsque l’on n’a pas d’a priori, c’est-à-dire lorsque l’on n’a pas d’hypothèse précise au sujet de la diffusion des protéines qu’on observe – une situation fréquente quand on s’intéresse à un système biologique encore peu exploré.
Une plateforme web pour l’analyse
Tout au long de ma thèse, j’ai échangé avec de nombreux biologistes, au sein de Sanofi et dans des laboratoires de recherche académique, afin de comprendre les outils qu’ils utilisent aujourd’hui pour analyser le mouvement des protéines auxquelles ils s’intéressent. J’ai pu constater que les méthodes varient grandement selon les domaines et les laboratoires, nécessitent plusieurs outils logiciels plus ou moins accessibles et faciles d’usage, ce qui nuit parfois à la portée des résultats obtenus et à leur reproductibilité. De plus, peu d’études estiment aujourd’hui la significativité statistique des résultats tirés d’analyses de trajectoire.
Après avoir éprouvé ma méthode sur des données provenant de plusieurs expériences différentes, de Sanofi et d’ailleurs, j’ai donc voulu la rendre accessible au plus grand nombre, via un outil d’analyse en ligne. J’ai développé pour cela un portail sur lequel les chercheurs peuvent déposer leurs trajectoires, les analyser avec mon algorithme et visualiser puis télécharger les résultats. À terme, j’espère que cette plate-forme permettra d’intégrer plusieurs autres types d’analyses, pour prendre en compte de nouveaux cas d’usage, et d’en partager les résultats, afin de permettre une plus grande transparence sur l’ensemble de l’analyse.
Conclusion
Observer le mouvement des protéines, c’est suivre le détail du fonctionnement de la machinerie cellulaire. Avec cette thèse, j’espère avoir contribué à la construction d’une méthodologie robuste qui permettra aux chercheurs de tirer les conclusions les plus exactes possibles de leurs observations, aussi bien pour la mise au point de médicaments que pour des expériences de biologie cellulaire.
Informations sur la thèse
J’ai réalisé ma thèse entre le laboratoire « Décision et processus Bayésien » dirigé par Jean-Baptiste Masson à l’institut Pasteur, et une équipe du département « Médecine de précision » de Sanofi, située sur le site de Vitry-sur-Seine. J’ai bénéficié pour cela d’un contrat CIFRE (Convention industrielle de formation par la recherche), une forme de collaboration qui permet aux laboratoires pharmaceutiques de nouer des liens avec la recherche académique. La thèse a été soutenue le 4 octobre 2022 devant un jury composé de :
- BERRY Hugues, Directeur de recherche INRIA
- PRESSE Steve,
- ALEXANDROU Antigoni,
- ALLASSONNIERE Stéphanie,
- Mme EL KAROUI Meriem,
- FRANÇOIS Paul,
- LOUPE Gilles,
- SAUER Markus,
- CASSE Alhassan,
- MASSON Jean-Baptiste
Informations sur le laboratoire
Le laboratoire de Jean-Baptiste Masson vient d’être confirmé pour dix nouvelles années à l’Institut. On y étudie les moyens sélectionnés par l’évolution pour servir de méthodes de décision aux organismes biologiques. Plusieurs doctorants, post-doctorants, ingénieurs de recherche et chercheurs permanents (dont des polytechniciens) s’intéressent pour cela, avec plusieurs approches, au comportement de la larve de mouche – un des rares organismes dont la structure de connexion des neurones entre eux soit entièrement connue.
Cela permet de mieux comprendre les liens entre les stimuli auxquels sont soumis les larves, l’activation de leurs neurones, la réponse de leur système moteur et in fine l’influence sur leur comportement. D’autres travaillent sur un dispositif de réalité virtuelle qui permet aux médecins de visualiser des scanners en trois dimensions afin de mieux préparer certaines opérations, ou d’affiner des diagnostics. Le laboratoire collabore sur certains sujets avec la start-up Avatar Medical, cofondée par Jean-Baptiste Masson et un de ses anciens étudiants.
Articles similaires :
 Finance quantitative à l’échelle de la microstructure : trading algorithmique et régulation
Finance quantitative à l’échelle de la microstructure : trading algorithmique et régulation
 Observation et modélisation des événements de pollution au Chili
Observation et modélisation des événements de pollution au Chili
 Mécanique pulmonaire personnalisée : modélisation, estimation et application à la fibrose pulmonaire
Mécanique pulmonaire personnalisée : modélisation, estimation et application à la fibrose pulmonaire
 Biomédecine et physique : Mouvements collectifs de micronageurs confinés
Biomédecine et physique : Mouvements collectifs de micronageurs confinés
 Contrôler l’aléatoire du vide grâce aux fluctuations quantiques
Contrôler l’aléatoire du vide grâce aux fluctuations quantiques